




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
碱茅 PutSnRK2基因表达、克隆及其蛋白纯化
[刘伟, 张欣欣 ]
]
]
|
|


第一作者:刘伟(1989-),男,河北唐山人,在读硕士生,研究方向为植物逆境分子生物学。E-mail:[email protected]
从NaHCO3逆境胁迫下碱茅( Puccinellia tenuiflora)的cDNA文库中克隆得到 PutSnRK2基因全长cDNA。 PutSnRK2基因序列中存在1 077 bp的开放阅读框,编码358个氨基酸,预测蛋白分子量为41.11 kDa,等电点为5.63。实时荧光定量PCR分析表明,在NaHCO3、NaCl、ABA和PEG胁迫下, PutSnRK2基因在碱茅叶、根中均受到显著诱导( P<0.05)。同时用pGEX-6p-3载体构建了 PutSnRK2基因的原核表达载体,将其转化大肠杆菌 BL21(DE3),经诱导表达纯化得到GST- PutSnRK2融合蛋白,为后续试验分析PutSnRK2蛋白的激酶活性、验证蛋白间的相互作用等研究奠定了基础。
In the present study, the full length cDNA of PutSnRK2 gene was cloned from the cDNA library of Puccinellia tenuiflora under the NaHCO3 stress. PutSnRK2 gene contains an open reading frame (ORF) of 1 077 bp, encodes 358 deduced amino acid residues (AARs) with a calculated molecular mass of 41.1 kDa and predicted pI of 5.63. Quantitative real-time PCR (qRT-PCR) analysis showed that the expression of PutSnRK2 gene was significantly induced ( P<0.05) in leaves and roots of the P. tenuiflora under stress of NaHCO3, NaCl, ABA, and PEG. At the same time, the prokaryotic expression plasmid of pGEX-6p-3- PutSnRK2 was constructed and transformed into Escherichia coli BL21 (DE3), and GST- PutSnRK2 fusion protein was induced and purified. These results provide foundation for further analyzing the activity of PutSnRK2 and protein-protein interaction.
蔗糖非酵解型蛋白激酶2 [SNF1(sucrose non-fermenting-1)-related protein kinase 2, SnRK2]是植物特有的一类Ser/Thr类蛋白激酶, 蛋白激酶SnRK2家族成员受渗透胁迫激活, 在脱落酸(abscisic acid, ABA)信号转导途径中有重要作用[1]。在拟南芥(Arabidopsis thaliana)中已经确定了10个SnRK2, 它们中的9个被高渗和盐胁迫激活, 并且这9个中的5个被ABA激活, 但都不受冷胁迫诱导[2]。过表达AtSnRK2.8、AtSnRK2C后能增强拟南芥的抗旱性[3]。拟南芥SnRK2.6、SnRK2E、OST1和蚕豆(Vicia faba)ABA-activated serine-threonine protein kinase (AAPK)被ABA激活, 它们都参与ABA调节的气孔关闭和ABA调节的基因表达[4, 5, 6]。在水稻(Oryza sativa)中, 10个成员已经被鉴定并且命名为SAPK1-10。它们都可以被高渗胁迫激活, 并且SAPK8-10被ABA激活[7]。过表达SAPK4能显著增强水稻的抗盐性[8]。在玉米中, 10个SnRK2已经被克隆, 大多数玉米SnRK2基因能被一种或更多非生物胁迫诱导[9]。在大豆(Glycine max)中, 鉴定出4个SnRK2成员, 都可以被高渗胁迫诱导[10, 11]。在小麦中, 只有一个SnRK2成员PKABA1可以被高渗胁迫和ABA诱导。并且在大麦糊粉层中瞬时过表达时抑制赤霉素诱导型启动子的活性[12, 13, 14, 15]。以往的研究结果表明, 许多SnRK2成员参与对环境刺激的反应。不同成员具有不同的表达模式, 这表明它们可能在非生物逆境胁迫应答中扮演不同的角色。
碱茅(Puccinellia tenuiflora)是一种禾本科单子叶盐生植物, 主要分布在我国的东北松嫩盐碱草甸草原上。与其它盐生植物相比, 它在极端盐恶劣的盐碱环境下(pH 9~10)可以完成其整个生命周期。且具有一套独特的适应盐碱逆境的生理机制, 同时也具有潜在的实用价值[16]。因此, 碱茅是一种理想的研究耐盐机制的盐生植物。本研究对PutSnRK2基因的生物信息学以及在非生物胁迫下的表达量进行分析, 有助于揭示PutSnRK2基因在非生物胁迫下的表达以及在调控植物生长发育中所起到的作用, 为进一步研究和利用基因工程技术培育抗盐碱作物提供理论依据。另外, 原核表达并纯化了的GST-PutSnRK2蛋白, 可为继续开展其激酶活性、验证蛋白相互作用等研究提供材料, 从而为进一步阐明PutSnRK2在响应其它非生物胁迫时的功能机制奠定基础。
1.1.1 植物材料 碱茅种子采自东北林业大学盐碱地生物资源环境研究中心安达野外实验基地。25 ℃室温, 相对湿度60%, 光照6 000 lx, 光照时间为16 h/8 h条件下, 水培养3周的碱茅幼苗为试材。
1.1.2 菌株和质粒 pMD18-T快速连接试剂盒购自TaKaRa公司; pGEX-6p-3载体购自GE公司。试验所需菌株大肠杆菌 JM109和BL21(DE3)来自本实验室。
1.1.3 试剂 Trizol试剂购自Invitrogen公司; 实时定量染料购自北京全式金公司; Glutathione SepharoseTM 4B购自GE公司; 质粒提取试剂盒购自TaKaRa公司; PureLink® Quick Gel Extraction Kit、restriction enzymes、ExpressLinkTMT4 DNA Ligase、低分子量蛋白Marker购自Thermo Fisher Scientific公司; GST抗体、碱性磷酸酶标记的山羊抗小鼠IgG(H+L)抗体购自碧云天公司。引物合成及测序由华大基因完成。试验中所用到的化学药品和培养基等试剂均为国产或进口分析纯。
1.2.1 碱茅总RNA的提取和cDNA第一链的合成 用Trizol试剂法提取碱茅总RNA[17]。用TaKaRa公司的PrimeScriptTMRT Reagent Kit(Perfect Real Time)反转录试剂盒反转录为cDNA。具体操作步骤如下:在无RNA酶的0.5 mL Ep管中加入2 μ L 5× PrimeScript Buffer(for Real Time), 0.5 μ L PrimeScript RT Enzyme Mix I, 0.5 μ L Oligo dT Primer(50 μ mol· L-1), 0.5 μ L Random 6 mers(100 μ mol· L-1), 500 ng Total RNA, 用RNase-free的ddH2O补足至10 μ L, 将混合液至于37 ℃ PCR仪上15 min, 而后85 ℃ 5 s终止反应。产物保存于-20 ℃冰箱中备用。
1.2.2 PutSnRK2基因的生物信息学分析 利用差异显示法, 在NaHCO3逆境下, 从碱茅cDNA文库中克隆得到碱茅逆境诱导蛋白激酶PutSnRK2基因全长cDNA[18]。该cDNA全长为1 726 bp。利用在线ORF finder软件(http://www.ncbi.nlm.nih.gov/gorf/gorf.html)分析PutSnRK2基因的ORF; 利用DNAMAN软件进行序列比对和分析; 利用在线ExPASy Compute pI/Mw tool软件对PutSnRK2蛋白进行等电点及分子量预测; 利用MEGA6.0软件进行多序列比对, 再用邻接算法(Neighbor-Joining)构建了系统进化树, 其中Bootstrap值设为1 000重复; 利用NCBI-BLAST及DNAMAN软件进行多重比对分析。
1.2.3 PutSnRK2基因在非生物胁迫下的表达特性分析 用实时荧光定量PCR来分析PutSnRK2基因在不同非生物胁迫下的相对表达水平, 具体方法如下:4个处理分别选取30粒种子, 每个处理3次重复, 以25 ℃室温, 相对湿度60%, 光照6 000 lx, 光照/黑暗时间为16 h/8 h, 水培养3周的碱茅幼苗为试材[19]。分别经300 mmol· L-1NaCl、150 mmol· L-1 NaHCO3、50 μ mol· L-1 ABA、10% PEG6000处理0、6、12、24 h后, 收集叶部组织、根部组织, 液氮速冻后迅速保存于-80 ℃超低温冰箱[20]。碱茅total RNA的提取及cDNA的合成同1.2。根据引物设计原则设计实时荧光定量PCR引物, 引物由Primer Premier 5软件设计, 序列上游:5’ -ACTACAAGAGAGACAACAGC-3’ , 下游:5’ -GTGGACGAAGGAGGTGGTTT-3’ ; 内参基因选用碱茅PutTublin基因, 序列上游:5’ -GGTAACATTGTGCTCAGTGGTGG-3’ , 下游:5’ -AACGACCTTAATCTTCATGCTGC-3’ , 反应在Mx3000P型荧光定量PCR仪上进行, 采用SYBR Green I染料法进行相对荧光定量检测。反应体系:2× TransStart Tip Green qPCR SuperMix 10 μ L, Primer上游(10 μ mol· L-1) 0.4 μ L, 下游(10 μ mol· L-1) 0.4 μ L, cDNA 1 μ L, Passive Reference Dye 0.4 μ L, ddH2O补足20 μ L。反应条件:94 ℃预变性30 s, 94 ℃变性5 s, 55 ℃退火15 s, 72 ℃延伸10 s, 40个循环, 最后根据反应的溶解曲线来判断扩增结果是否特异。每个反应3个平行, 每次试验制备不含cDNA的阴性样品, 得到的Ct值用2-Δ Δ Ct法来计算基因的相对表达水平。
1.2.4 PutSnRK2基因原核表达载体的构建及目的蛋白的表达与纯化 碱茅的cDNA文库(150 mmol· L-1 NaHCO3处理)由本实验室构建完成。用文库载体通用引物扩增碱茅PutSnRK2基因全长片段。上游为:5’ -ACTGCTCCTCAGTGGATGTT-3’ , 下游为:5’ -CCCTCACTAAAGGGAGATCC-3’ 。反应条件为:94 ℃预变性2 min, 94 ℃变性30 s, 56 ℃退火30 s, 72 ℃延伸120 s, 30个循环后, 72 ℃延伸10 min。反应体系:TaKaRa Ex Taq(5 U· μ L-1) 0.25 μ L, 10× Ex Taq Buffer 5 μ L, dNTP Mixture(2.5 mmol· L-1) 4 μ L, Template 1 μ L, 上游引物(10 μ mol· L-1) 1 μ L, 下游引物 (10 μ mol· L-1) 1 μ L, ddH2O补足至50 μ L。纯化后的PCR产物连接pMD18-T载体。为了将PutSnRK2基因构建到原核表达载体pGEX-6p-3的EcoRⅠ 和XhoⅠ 酶切位点之间, 在上下游引物中分别设计有EcoRⅠ 和XhoⅠ 识别切割位点。引物由Primer Premier 5软件设计, 序列上游:5’ -GAATTCGATGGAGAGGTACG-3’ , 下游:5’ -CTCGAGTCAGGTGATGTGGA-3’ ; 以实验室保存的连接到pMD18-T载体上的PutSnRK2基因为模版对PutSnRK2进行扩增。反应条件为:94 ℃预变性2 min, 94 ℃变性30 s, 54 ℃退火30 s, 72 ℃延伸90 s, 30个循环后, 72 ℃延伸10 min。反应体系:TaKaRa Ex Taq(5 U· μ L-1) 0.25 μ L, 10× Ex Taq Buffer 5 μ L, dNTP Mixture(2.5 mmol· L-1) 4 μ L, Template 1 μ L, 上游引物(10 μ mol· L-1) 1 μ L, 下游引物 (10 μ mol· L-1) 1 μ L, ddH2O 补足至50 μ L。将PCR产物电泳并回收目的条带, 然后将纯化后的PutSnRK2连接pMD18-T载体, 连接产物转化大肠杆菌JM109并过夜培养。将提质粒酶切鉴定的阳性克隆送华大基因测序。测序正确的带酶切位点的PutSnRK2基因和pGEX-6p-3质粒用EcoRⅠ 和XhoⅠ 双酶切后电泳回收目的片段和载体片段, ExpressLinkTM T4 DNA Ligase连接过夜, 转化大肠杆菌JM109, 氨苄青霉素筛选阳性克隆, 提取质粒, 进行酶切鉴定, 正确的重组质粒进行下游试验。
将测序正确的重组质粒转化大肠杆菌BL21(DE3), 挑取阳性克隆接种于LB液体培养基中(含Amp 100 μ g· L-1), 37 ℃震荡培养过夜。次日按1%比例转接后, 37 ℃震荡培养使OD600 nm达到0.5后, 加入IPTG至终浓度1 mmol· L-1, 37 ℃诱导0、15、30、60、120和210 min, 并离心收集每个时间的菌体, 进行10% SDS-PAGE鉴定[21]。
将含有pGEX-6p-3-PutSnRK2质粒的大肠杆菌过夜培养物加入到200 mL液体LB培养基中, 25 ℃振荡培养。当菌液的OD600 nm达到0.5时, 取700 μ L菌液, 离心收集菌体, 冻入-80 ℃冰箱中。其他菌液加入IPTG 使其终浓度为0.5 mmol· L-1, 依然25 ℃振荡培养, 6 h后取700 μ L菌液, 离心收集菌体, 冻入-80 ℃冰箱中。其他菌液离心收集菌体后加入裂菌缓冲液并进行超声波碎菌, 随后收集上清进行纯化。具体纯化操作参照Glutathione SepharoseTM 4B纯化试剂说明书。Western Blotting鉴定, 方法参照文献[22]及碧云天GST抗体、碱性磷酸酶标记的羊抗鼠IgG(H+L)抗体说明书。
试验数据分析采用Mx 3000P型荧光定量PCR系统软件和Excel 2007软件进行数据录入、计算、做图; 采用SPSS 17.0统计软件中AVONA对PutSnRK2表达量数据进行方差分析, LSD多重比较法分析处理样品间差异, 显著水平为0.05。
对PutSnRK2进行ORF分析, PutSnRK2基因序列中的开放阅读框大小为1 077 bp, 编码358个氨基酸。利用ExPASy Compute pI/Mw tool软件预测PutSnRK2蛋白等电点为5.63, 分子量为41.11 kDa。搜索SnRK2家族已克隆基因的氨基酸序列, 用DNAMAN软件进行氨基酸序列比对并构建系统进化树。系统进化树表明, PutSnRK2蛋白与拟南芥、水稻等作物聚类在一起, 系统进化亲缘关系在76%左右(图1)。碱茅是禾本科的植物, 系统进化树分析结果也表明它与水稻亲缘关系较近。其中PutSnRK2与水稻SAPK7有很高的相似性, 同属于SnRK2b亚家族[18]。运用DNAman软件将随机挑选的水稻SAPK进行氨基酸多重性比较(图2), 比较结果显示, 碱茅与水稻的SnRK2相比较蛋白的N-末端是相对保守的, 并且在蛋白激酶的结合区域和丝氨酸、苏氨酸蛋白激酶活性位点十分保守; C-末端较为多样。
 | 图1 PutSnRK2蛋白与拟南芥、水稻同源蛋白的系统进化树分析Fig.1 Phylogenetic analysis of PutSnRK2 protein with Arabidopsis thaliana, Oryza sativa protein |
以碱茅中的Tublin为内参基因, 通过对PutSnRK2基因在几种非生物胁迫下表达的情况的实时荧光定量PCR的分析发现(图3), 碱茅叶、根中PutSnRK2在300 mmol· L-1 NaCl、150 mmol· L-1NaHCO3、50 μ mol· L-1 ABA和10% PEG胁迫下均受到了显著(P< 0.05)的诱导。在叶中, PutSnRK2在NaCl、NaHCO3、ABA、PEG胁迫下显著(P< 0.05)下调。其中在NaHCO3胁迫24 h时, 叶中下调了20%左右; 而在NaCl、ABA、PEG胁迫24 h时, 其表达量已经下调了75%以上。在根中, PutSnRK2在NaCl、NaHCO3、ABA、PEG胁迫下表达均显著上调(P< 0.05)。其中在NaHCO3胁迫6 h时, 根中PutSnRK2表达量已到最高, 上调了3倍左右; 在NaCl、ABA、PEG胁迫12 h时, 表达量达到最高, 上调1.5~2.5倍。
 | 图2 PutSnRK2与水稻蛋白的多重比对分析Fig.2 Multiple sequences alignment of PutSnRK2 with Oryza sativa protein注:蛋白激酶结合区和丝氨酸苏氨酸蛋白激酶活性位点分别在红框和緑框内表示。保守的ATP结合位点(K, 赖氨酸残基)和活性位点(D, 天冬氨酸残基)分别用红色的三角形和绿色的五角星表示。Note: Protein kinases ATP-binding region signature and serine/threonine protein kinases active-site signature are indicated by a red box and a green box, respectively. The conserved ATP binding site (K, lysine residue) and the active site (D, aspartic acid residue) are marked with a red triangle and a green pentacle, respectively. |
 | 图3 PutSnRK2基因在非生物胁迫下的表达特性Fig.3 Expression characteristic of PutSnRK2 under abiotic stress注:* 表示各处理与对照间差异显著(P< 0.05)。Note: * indicated significant difference between the treatment and control at 0.05 level. |
采用设计的引物通过PCR扩增获得带有酶切位点的PutSnRK2基因(图4A)。连接T载体后, 转化大肠杆菌JM109, 对酶切鉴定的阳性克隆测序。分别将连接到T载体上的PutSnRK2与提取的质粒pGEX-6p-3用EcoRⅠ 和XhoⅠ 进行双酶切、胶回收。再用ExpressLinkTM T4 DNA Ligase进行连接, 转化大肠杆菌JM109, 将过夜培养后筛选到的阳性克隆摇菌, 用质粒小提试剂盒提取质粒并对其进行双酶切验证(图4B), 得到重组质粒。
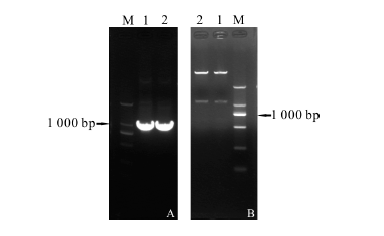 | 图4 PutSnRK2基因的克隆和双酶切验证重组质粒Fig.4 Cloning of PutSnRK2 gene from Puccinellia tenuiflora and double restriction enzymes digestion and verification of recombinant vector注:M为DNA Marker, A(1, 2)为扩增产物; B(1, 2)为pGEX-6p-3-PutSnRK2。Note:M, DNA Marker; A(1, 2), amplification products; B(1, 2), pGEX-6p-3-PutSnRK2). |
将重组质粒pGEX-6p-3-PutSnRK2转化BL21(DE3)感受态细胞, 过夜培养后摇菌, 吸取1 μ L的菌液作为模版进行菌液PCR并进行琼脂糖凝胶电泳, 挑选鉴定正确的阳性克隆进行小量诱导表达, SDS-PAGE电泳结果显示(图5A), 诱导出分子量大约为60 kDa的蛋白。其中GST标签分子量约为26 kDa, 目标蛋白预测分子量为41.1 kDa, 与预测的融合蛋白67 kDa偏小。对融合蛋白进行总平均疏水指数分析(图6)。进行大量诱导后结果表明:GST-PutSnRK2融
合蛋白是可溶蛋白, 并且在上清中表达。大量诱导及纯化后得到的蛋白质(图5B), 经过Western Blotting检测 (图5C)有信号, 表明成功纯化得到了原核表达的纯化蛋白GST-PutSnRK2。
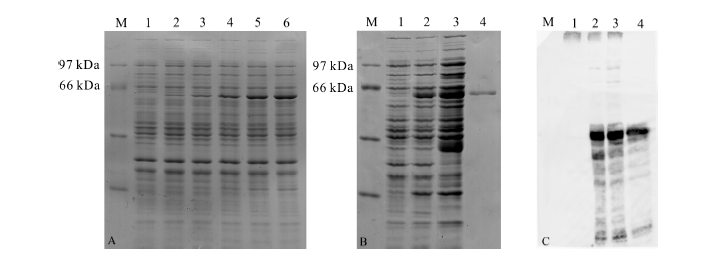 | 图5 GST-PutSnRK2蛋白的诱导与纯化Fig.5 Induction and purification of GST-PutSnRK2 ProteinA:M, 低分子量蛋白Marker; 泳道1-6, 分别为IPTG诱导0、15、30、60、120、210 min的BL21总蛋白); B、C:M, 低分子量蛋白Marker; 1, 诱导0 min的BL21总蛋白; 2, 诱导6 h的BL21总蛋白; 3, 诱导6 h的BL21上清; 4, 纯化的GST-PutSnRK2蛋白)。A: M, low molecular weight standard proteins marker; Lane 1-6, the total protein of BL21, induced in 0, 15, 30, 60, 120, 210 min by IPTG; B, C: M, low molecular weight standard proteins marker; Lane 1, the total protein of BL21, induced 0 min; Lane 2, the total protein of BL21, induced 6 h; Lane 3, the supernatant of BL21, induced 6 h; Lane 4, the purification of GST-PutSnRK2. |
 | 图6 GST-PutSnRK2融合蛋白的总平均疏水指数分析Fig.6 Grand average of hydropathicity analysis of GST-PutSnRK2 Protein |
导致植物生长发育缓慢的主要非生物胁迫因素有干旱、盐碱和低温等。而我国盐碱地面积逐渐增加, 土壤盐害严重影响作物的生长发育和产量提高, 因此, 克隆盐胁迫应答相关基因, 研究耐盐机理, 对于培育耐盐品种(系)、提高盐胁迫下作物产量有重要的意义。碱茅因为有一套独特的生理机制使它在盐碱逆境下能比其它物种更好的生长发育。因此, 它对于研究耐盐机制的研究具有潜在的使用价值。SnRK2基因家族在植物多种信号传导和抗逆过程中发挥重要作用[23]。系统进化分析(图1)和氨基酸多重比对(图2)发现SnRK2蛋白激酶是一类非常保守的蛋白, 结果显示碱茅与水稻的SnRK2比较蛋白的N-末端是相对保守的, 并且在蛋白激酶的结合区域和丝氨酸、苏氨酸蛋白激酶活性位点十分保守; C-末端较为多样。此结果表明, SnRK2蛋白N-末端的保守区是响应胁迫的重要部分[18], 而C-末端可能与不同胁迫下产生不同表达模式有关。
大量研究表明, SnRK2蛋白激酶影响植物的生长发育, 并且在响应各种非生物胁迫中起重要作用[2, 7, 9, 23, 24, 25, 26]。本研究发现PutSnRK2基因在转录水平上受到NaCl、NaHCO3、ABA和PEG等多种非生物胁迫的诱导。在叶中PutSnRK2表达均受到显著抑制, 在根中表达均显著上调, 说明它可能在响应非生物胁迫过程中起一定的作用。在NaCl和NaHCO3胁迫下根的表达量较高, 显示出对盐碱胁迫有较强的响应。但是, 其逆境胁迫下的抗逆相关功能需进一步验证。此研究为以后构建转基因株系探究碱茅SnRK2基因在非生物胁迫下的确切功能提供参考。
获得大量高纯度的蛋白质是对蛋白质进行生物化学分析的基础, 在原核表达蛋白系统中比较容易快速获取大量高纯度的蛋白质, 且有多种亲和纯化标签可以选择, 因此原核表达蛋白被广泛地用于表达真核生物的蛋白质[27]。本研究将PutSnRK2基因构建到pGEX-6p-3原核表达载体上, 并转入BL21(DE3)中。结果表明, 诱导的融合蛋白比预测值偏小, 利用ExPASyProtParam tool软件(http://web.expasy.org/protparam/)对融合蛋白分析, 它的亲水性平均系数为-0.462, 疏水性低, 导致泳动速度偏快, 电泳结果比预测值偏小。对于GST-PutSnRK2融合蛋白, 用0.5 mmol· L-1 IPTG、25 ℃诱导6 h, 获得的蛋白质经过还原型谷光氨肽的洗脱, 收集到有足够纯度和足够量的GST-PutSnRK2融合蛋白。这种方法为通过生物化学方法分析PutSnRK2的激酶活性、验证蛋白间的相互作用等功能, 深入研究PutSnRK2提供材料。
本研究发现, PutSnRK2具备植物SnRK2基因家族的固有特征, 同时RT-PCR分析表明其能够响应盐、碱、ABA和干旱等非生物胁迫, 说明它可能在响应非生物胁迫过程中起一定的作用。同时, 本研究得到了纯化的PutSnRK2蛋白, 为分析它的功能提供了材料。要揭示碱茅SnRK2基因在非生物逆境胁迫中的确切功能尚需要借助转基因植物等手段开展大量深入细致的研究, 目前有关工作正在进行中。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|